
RESEARCH PROJECTS
See a full list of my publications on Google Scholar.
Active Projects
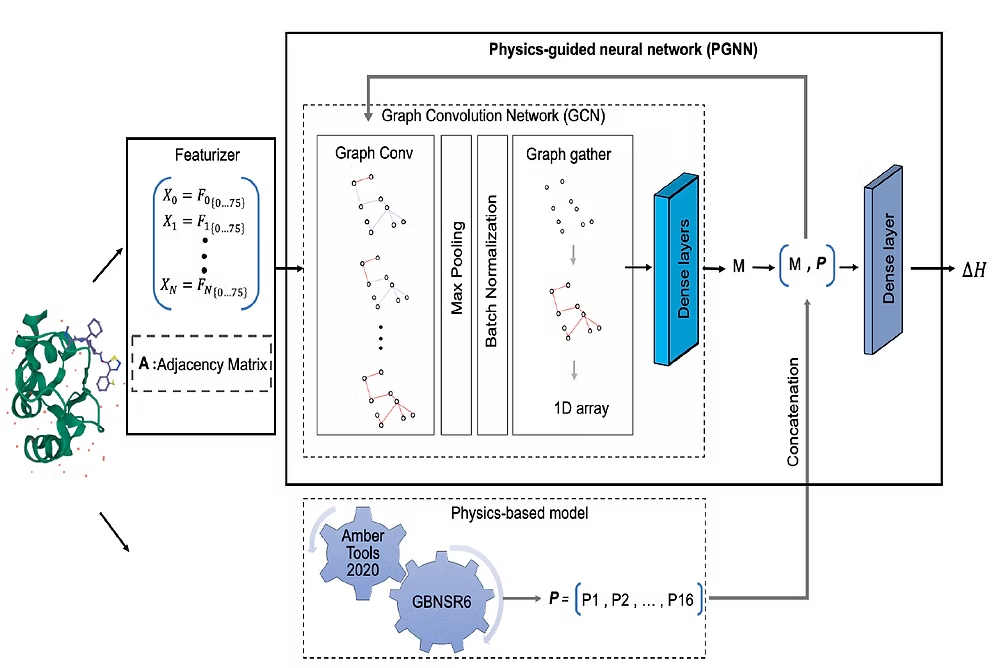
Development of a physics-based deep learning network to calculate binding free energies of small protein–ligand complexes in an implicit solvent model. We build our model based on a deep learning framework called Deepchem, a Python library mainly used in drug discovery processes.
Publications:
- Calculation of Protein-Ligand Binding Entropies Using a Rule-based Molecular Fingerprint, Elsevier 2024 (LINK)
- Calculating the Binding Entropy of Host–Guest Systems with Physics-Guided Neural Networks. IEEE-BIBM 2022 (LINK)
- A Physics-Guided Neural Network for Predicting Protein–Ligand Binding Free Energy. Biomolecules 2022 (LINK)
- Calculation of Protein–Ligand Binding Free Energy Using a Physics-Guided Neural Network, IEEE-BIBM 2021 (LINK)
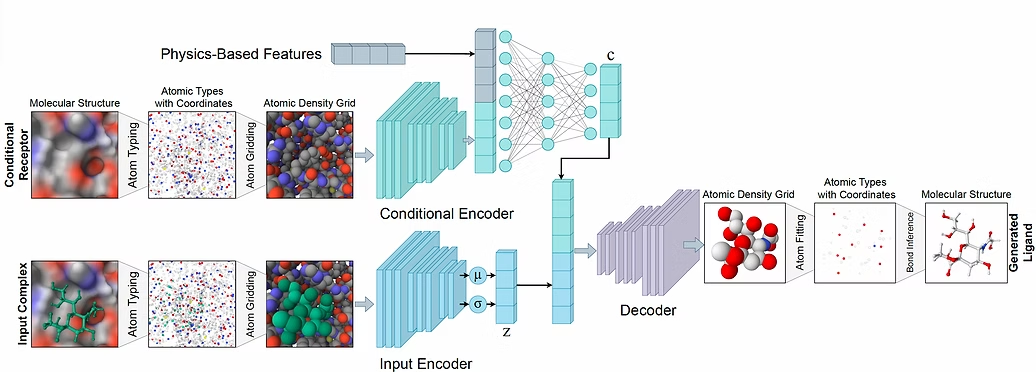
Deep generative models have recently been introduced for sampling the chemical space and discovering new ligands. Unlike experimental methods, the computational pipeline enables researchers to modify the generative algorithm quickly and effortlessly test the results from different perspectives. This paradigm shift opens up new opportunities to create a rich database of artificial structures and evaluate their synthesizability and viability.
Publications:
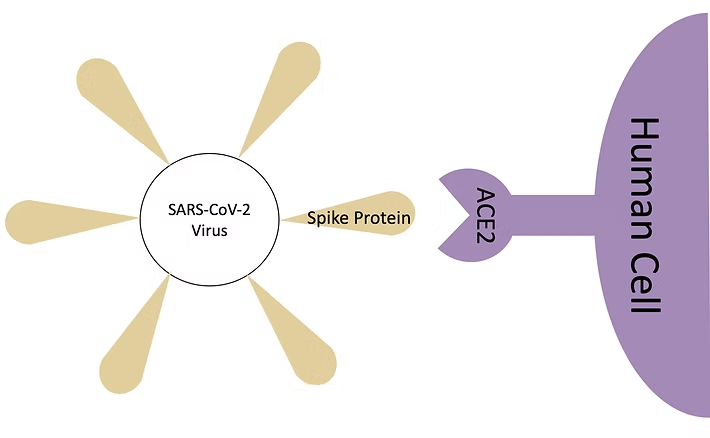
Evaluation of the potential of a molecular mechanics generalized Born surface area (MM/GBSA) approach to estimate the binding free energy between the SARS-CoV-2 spike receptor-binding domain (wild type and mutants) and the human ACE2 receptor.
Publications:
- An Effective MM/GBSA Protocol for Absolute Binding Free Energy Calculations: A Case Study on SARS-CoV-2 Spike Protein and the Human ACE2 Receptor, Molecules 2021 (LINK)
- Binding Free Energy of the Novel Coronavirus Spike Protein and the Human ACE2 Receptor: An MMGB/SA Computational Study, ACM-BCB 2020 (LINK)
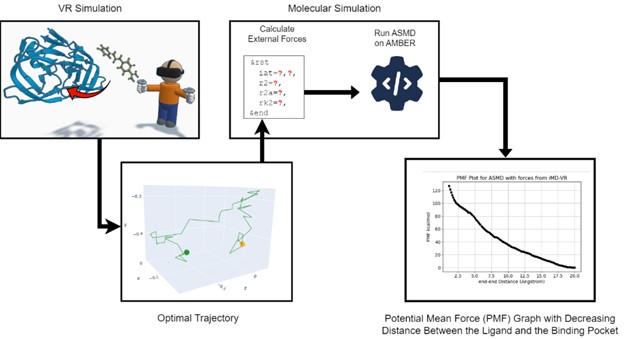
Adaptive steered molecular dynamics (ASMD) is a computational biophysics method in which an external force is applied to a selected set of atoms or a specific reaction coordinate to induce a particular molecular motion. This research proposes a novel method to guide ASMD with optimal trajectories collected from human experiences through experiments in virtual reality.
Publications:
- Molecular Docking Improved With Human Spatial Perception Using Virtual Reality, IEEE 2024 (LINK)
Past Projects
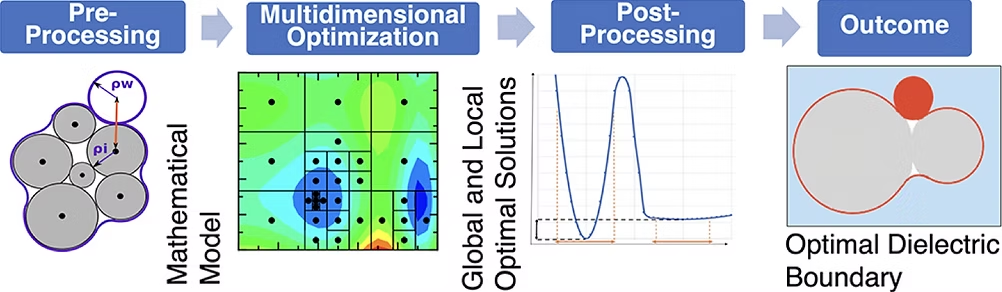
Optimization of atomic radii in an implicit solvent model to obtain close agreement with experimental results in terms of calculating electrostatic binding free energies. The underlying massively parallel optimization method, VTDIRECT95, runs on Virginia Tech clusters.
Publications:
- Optimal Dielectric Boundary for Binding Free Energy Estimates in the Implicit Solvent, JCIM 2024 (LINK)
- Multidimensional Global Optimization and Robustness Analysis in the Context of Protein-Ligand Binding, JCTC 2020 (LINK)
- Robustness of Multidimensional Optimization Outcomes: A General Approach and a Case Study, SpringSim 2020 (LINK)
- Grid-based surface generalized Born model for calculation of electrostatic binding free energies, JCIM 2017 (LINK)
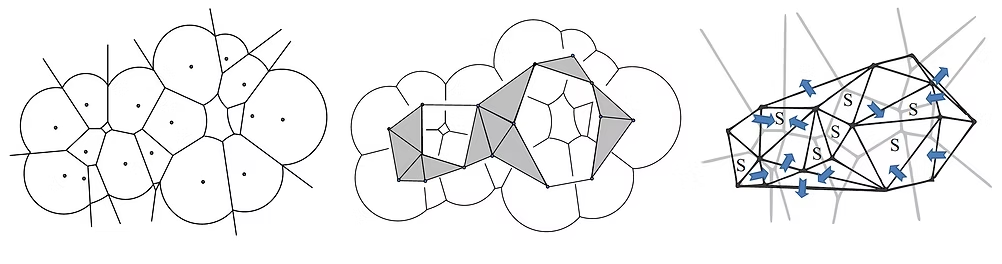
Introduction of a novel geometrical metric to identify protein binding pockets. The proposed metric is based on the von Neumann entropy of the weighted Delaunay triangulation of protein pockets.
Publications:
- Structure-Based Analysis of Protein Binding Pockets Using Von Neumann Entropy, ISBRA 2014 (LINK)